 https://www.genetica.cz
+420 272 701 055
Služeb 4
Praha
https://www.genetica.cz
+420 272 701 055
Služeb 4
Praha

1.png)


.png)
.png)
Seer Proteograph
Introduction
Proteins in biofluids like blood are predominantly composed of high-abundance proteins, such as albumins and globulins, which together account for over 90% of the total protein content. These abundant proteins often obscure low-abundance proteins that carry critical insights into disease mechanisms, biomarker discovery, and therapeutic targets. Traditional proteomic methods frequently struggle to detect these low-abundance proteins without extensive preprocessing, making it challenging to obtain meaningful data.
The Proteograph Product Suite addresses these challenges through Seer’s proprietary nanoparticle technology. These unique nanoparticles are designed to selectively bind proteins across a wide dynamic range, enriching low-abundance proteins without the need for complex preprocessing. By overcoming these barriers, the fully automated Proteograph XT workflow provides comprehensive, high-resolution proteomic analysis of blood and other biofluids, unlocking access to the "hidden" proteome with efficiency and reproducibility. The system’s seamless integration into laboratory workflows makes it a valuable tool for researchers seeking reliable proteomic data at scale.

Workflow
The Proteograph workflow consists of five key steps:
- Sample Preparation: Biological samples are processed to generate proteomic solutions suitable for nanoparticle labeling.
- Nanoparticle Labeling: Engineered nanoparticles bind specific subsets of proteins based on surface physicochemical properties.
- Elution and Digestion, Quantification: Proteins bound to the nanoparticles are eluted and digested into peptides and quantificated for their concentration. Afterwards, they are frozen until their reconstitution and analysis by LC-MS.
- LC-MS Analysis: The resulting peptides are analyzed using Liquid Chromatography-Mass Spectrometry (LC-MS) to provide high-resolution data. This is not provided directly by Seer Proteograph.
- Data Analysis: The Proteograph Analysis Suite (PAS) is a dedicated cloud-based software solution for processing, analyzing, and visualizing proteomics data generated by Seer workflow and liquid chromatography-mass spectrometry (LC-MS). The integrated search engines power rapid identification and annotation of LC-MS data and the variety of quality control tools.
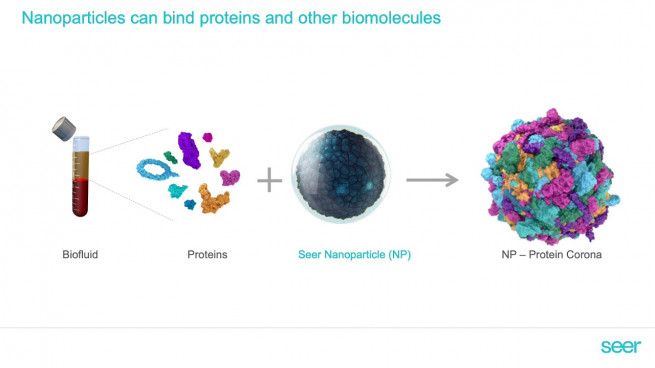
The Seer Proteograph sample preparation takes approximately two days.
Data Analysis and Results
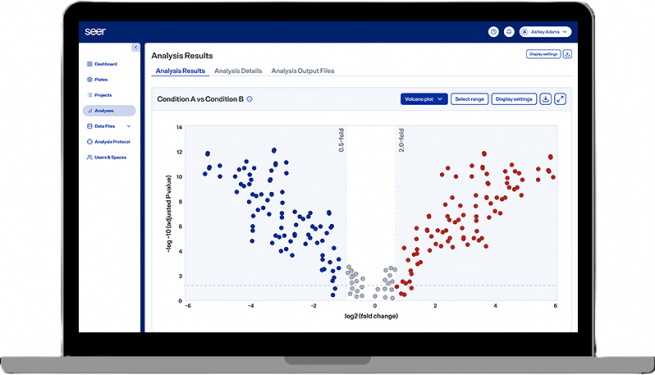
Seer Proteograph analysis is powered by the Proteograph Analysis Suite (PAS), a cloud-based software solution designed to simplify and enhance proteomic data analysis. Using an integrated search engine and Seer’s proprietary spectral library, PAS enables rapid peptide identification and protein quantification. Pre-configured pipelines streamline data filtering, including peptide annotation, false discovery filtering, and statistical protein assignment, making the system accessible even to users without advanced bioinformatics expertise. Results are presented in user-friendly formats, including interactive visualizations such as plate maps, protein group overlaps, and quality control metrics. Comparative analyses, such as differential protein expression or group comparisons, are supported by advanced visualization tools like intensity plots and protein-protein interaction networks. Additionally, PAS offers proteogenomics workflows, allowing integration with genomic variant data to identify peptide variants and explore their biological relevance.
Applications
- biomarker discovery
- disease mechanism studies
- drug development and response monitoring
- precision medicine research
- quantitative proteomics multi-omics studies
Validation of Seer Capabilities
In collaboration with the GeneTiCA Experience Centre, Prof. Zbyněk Zdráhal from CEITEC Brno, CZ, and Dr. Jana Jakubíková from the Cancer Research Institute, Slovak Academy of Sciences, Bratislava, Slovakia, we conducted a comparative study to evaluate the capabilities of Seer Proteograph against conventional sample processing methods. Seer Proteograph workflow identified nearly five times more proteins compared to traditional approaches, showcasing its unparalleled ability to overcome the challenges of plasma proteomics. To explore the full results of this study, view the detailed poster.
